By Caryn Lipson
KEY FINDINGS
- A saline placebo is the most rigorous vaccine trial design available, and still not enough to establish meaningful safety or real-world effectiveness.
- The vaccine prevented roughly one case of mild confirmed flu per 20 vaccinated children. Against the strains it was specifically designed to match, that number rises to one case per 48.
- The strain this trial succeeded against most clearly has not circulated globally since 2020 and has been removed from many vaccines. The strain it failed against is the only influenza B lineage still circulating.
- Five deaths in the vaccine arm and one in the placebo deemed not related to the vaccine. While the total is consistent with background mortality in the trial countries there is no explanation for the excess mortality in the vaccine arm.
Introduction
A saline-placebo controlled trial is widely assumed to provide a more accurate assessment of a vaccine’s safety and effectiveness than trials using an active comparator or antibody measurements alone. That assumption is reasonable as far as it goes. A saline-placebo trial is the most rigorous design available in vaccine research and genuinely rare in flu vaccine development. The majority of flu vaccines were approved on antibody levels alone, or compared against another vaccine rather than an inert control.
But more rigorous is not the same as sufficient. What follows is a close reading of a saline-placebo controlled trial, examining what it actually showed, what it did not and could not show, and where its key finding no longer applies.
The paper, “Efficacy, immunogenicity, and safety of a quadrivalent inactivated influenza vaccine in children aged 6-35 months” by Pepin et al. (2019), tested VaxigripTetra, a Sanofi quadrivalent influenza vaccine, in healthy children aged 6 to 35 months. VaxigripTetra is not marketed in the United States; it is approved in the European Union, South Korea, Taiwan, and other markets across Asia and Latin America, including Israel.1
About the Trial
- The manufacturer, Sanofi, was responsible for every stage of the trial: it funded it, designed it, ran the statistics in-house, paid the medical writer, decided to publish, and approved the final version. When a company runs its own trial, every judgment call (what to feature, how long to monitor safety, how to describe the results) is made by the party with the most to gain from a favorable outcome.
- The trial’s aim was to extend the vaccine’s approved age range downward. It was already licensed for children 3 and older; this trial was designed to support use in children aged 6 to 35 months.
- It included three vaccines: the quadrivalent vaccine being tested (two A strains and two B strains, B/Yamagata and B/Victoria lineages), and two existing licensed trivalent vaccines used as comparators, one containing B/Yamagata and one containing B/Victoria. Neither comparator had been tested against a saline placebo in children this age. Their licensure rested on immunogenicity bridging2 (demonstrating similar antibody responses to an already-approved product) rather than a placebo-controlled efficacy trial. Using either as a reference baseline therefore rests on the same assumption the trial was designed to go beyond.
- The trial used a saline placebo and ran from March 2014 to July 2016 over four influenza seasons.
- The investigators called the trial’s “representativeness” a major strength because it was “conducted over a wide geographical area in both hemispheres.” However, per-country enrollment (which appears only in the registry, not the paper) shows that about half the 5,805 children (2,999) were recruited in the Philippines. A child growing up there has a different nutritional baseline, different infectious disease exposure, different immune history, and different genetic background than a child in Israel, the United States, or Europe. All of these factors affect how a vaccine performs and how the body responds to it. A trial conducted predominantly in one population tells you how the vaccine performed there. It does not tell you how it is likely to perform in a different population.
Regarding Efficacy
- The paper itself states that no “correlate of protection,” no antibody level that reliably signals protection, has been established in children this age. This means that raising antibodies is not the same as being protected. This matters because much of vaccine licensing rests on immunogenicity (antibody or titer levels) data alone. The clinical results (actual flu cases) are the only meaningful measure here, and they are what the table below reflects.
- The paper reported that the vaccine halved the incidence of flu among trial participants. That figure describes the relative reduction. The table below shows what the numbers look like in absolute terms, which is what actually matters for a parent making a decision:
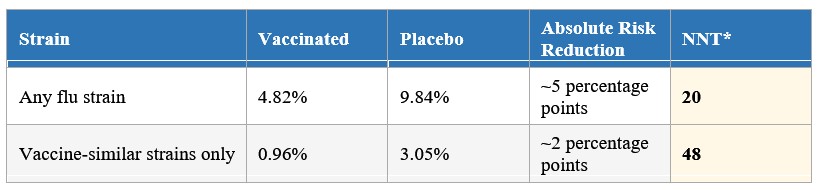
* NNT = Number Needed to Treat: the number of children who must be vaccinated to prevent one case of mild confirmed flu. Both figures apply to mild illness only, not hospitalization or serious disease. The matched-strain NNT of 48 is higher than the any-strain NNT of 20, which may seem counterintuitive. You would expect the vaccine to perform best against the strains it was specifically designed to match, and in relative terms it does (68.4% vs 51%). But the absolute benefit is smaller because matched-strain flu was rarer in this population during these seasons: the placebo attack rate for matched strains was only 3.05%, compared to 9.84% for any strain. Fewer cases to prevent means a smaller absolute reduction, regardless of how effective the vaccine is at preventing them. This is precisely why relative and absolute figures tell different stories, and why the relative headline can be misleading.
- The trial also illustrates how a conclusion can outrun its own data. Against the B/Victoria strain, the vaccine came up short: efficacy was not statistically demonstrated, and the antibody response was weaker than the comparator. The paper acknowledges this directly:
“Efficacy of IIV4 against the B/Victoria strain will have to be established in further studies.”
Yet the conclusion claims protection from both B strains regardless:
“By including a second B-lineage strain, IIV4 should provide additional protection beyond IIV3, irrespective of which B lineage circulates during a given season or region.”
- The strain the trial succeeded against most clearly (B/Yamagata) has not been detected globally since 2020 and has been removed from many vaccines. The strain it failed against (B/Victoria) is the only influenza B lineage still circulating.3
Regarding Safety
- The investigators concluded that the vaccine and placebo arms were similar except for injection-site reactions:
“…except for a higher proportion of participants reporting solicited injection-site reactions in the IIV4 group (39.9%) than in the placebo group (31.9%), proportions reporting solicited reactions and adverse events were similar for the IIV4, IIV3, and placebo groups.”
However, consider what “similar” actually means here, and what was and was not captured:
- How reactions were collected matters. Parents reported expected reactions at home from a fixed checklist for 7 days. For anything unexpected, a parent first had to notice something, decide it was serious enough to contact the trial site, and an investigator then had to decide it was worth recording. The data reflects how many events cleared those hurdles, not how many events occurred.
- The monitoring window was short. Serious adverse events were tracked for 6 months. No long-term follow-up was planned or conducted. A product given to infants and intended for annual use was never followed beyond half a year in this trial.
- The paper committed to monitoring eight serious conditions (including Guillain-Barre syndrome, anaphylaxis, and encephalitis) but reported only a combined total with no breakdown by condition.4 It cannot be determined from the paper or the registry whether any of those conditions actually occurred. Additionally, two events that appear in the regulatory registry are absent from the published paper: one case of Bell’s Palsy and one case of Kawasaki’s disease, both in the vaccine arm, both zero in the placebo.
- The trial showed no benefit for severe disease. Hospitalizations were equal, three in each arm, far too few to detect a difference either way. Hospitalization is the outcome that matters most for a parent making this decision, and the trial was never designed to address it.
- Five deaths in the vaccine arm, one in the placebo. The regulatory registry (the fuller record) shows five deaths in the vaccine arm and one in the placebo arm. The published paper reported only four, not five. Both arms enrolled similar numbers of healthy children, so the difference cannot be explained by one group being sicker. Sanofi judged none of the deaths vaccine-related but provided no explanation for the unequal distribution.5
Trial Stopped Midway
- The trial was stopped early once efficacy was demonstrated, at 5,806 children enrolled instead of the planned 8,536. Stopping a trial at its most favorable point is a known source of inflated efficacy estimates: the result captures the best-case snapshot rather than the full picture. Early stopping also meant the safety database was smaller than planned. Adverse events that may have appeared in the remaining enrollment were never captured.
What This Analysis Shows
A saline placebo is a legitimate standard to ask for. This trial used one, and that makes it presumably more rigorous than most flu vaccine research. What the analysis above shows is that a trial that looks solid from the outside, published in a peer-reviewed journal with a saline placebo, can still leave the most important questions unanswered. The difference between appearance and evidence is worth understanding before accepting any claim of safety and effectiveness as settled.
Footnotes